前提条件
准备数据文件,GATK测试中需要两类数据文件:参考基因组数据文件(FASTA格式文件)和原始测序数据(FASTQ或BAM格式文件)。
在本测试算例中使用的数据文件如下:
- 基因组数据文件(FASTA格式文件)
human_g1k_v37.fasta
以下两个vcf来自于千人基因组和Mills项目,里面记录了那些项目中检测到的人群Indel区域。- Mills_and_1000G_gold_standard.indels.hg19.sites.vcf.gz
- 1000G_phase1.indels.hg19.sites.vcf.gz
以下文件汇集的是目前几乎所有的公开人群变异数据集。
dbsnp132_20101103.vcf
变异数据集根据测序目的选择一到三种数据。
- 原始测序数据(FASTQ或BAM格式文件)
- SRR742200_1.fastq
- SRR742200_2.fastq
- 使用PuTTY工具,以root用户登录服务器。
- 执行以下命令创建“input”文件夹。
mkdir projectDir cd projectDir mkdir input
- 执行以下命令解压算例文件。
gzip -d SRR742200_1.fastq.gz gzip -d SRR742200_1.fastq.gz gzip -d human_g1k_v37.fasta.gz gzip -d dbsnp132_20101103.vcf.gz
- 执行以下命令使用blat生成“2bit”格式文件。
faToTwoBit human_g1k_v37.fasta human_g1k_v37.fasta.2bit
- 执行以下命令将所有数据文件放进“input”文件夹内。
cp human_g1k_v37.fasta human_g1k_v37.fasta.2bit SRR742200_1.fastq SRR742200_2.fastq dbsnp132_20101103.vcf input
操作步骤
- 使用PuTTY工具,以root用户登录服务器。
- 执行以下命令,BWA中“index”对“fasta”文件构建索引。
bwa index -a bwtsw human_g1k_v37.fasta
回显显示如下信息:
[bwa_index] Pack FASTA... 27.89 sec [bwa_index] Construct BWT for the packed sequence... [BWTIncCreate] textLength=6203609478, availableWord=448508744 [BWTIncConstructFromPacked] 10 iterations done. 99999990 characters processed. [BWTIncConstructFromPacked] 20 iterations done. 199999990 characters processed. [BWTIncConstructFromPacked] 30 iterations done. 299999990 characters processed. [BWTIncConstructFromPacked] 40 iterations done. 399999990 characters processed. [BWTIncConstructFromPacked] 50 iterations done. 499999990 characters processed. [BWTIncConstructFromPacked] 60 iterations done. 599999990 characters processed. [BWTIncConstructFromPacked] 70 iterations done. 699999990 characters processed. [BWTIncConstructFromPacked] 80 iterations done. 799999990 characters processed. [BWTIncConstructFromPacked] 90 iterations done. 899999990 characters processed. <以下输出内容省略>
- 执行以下命令,查看生成索引文件。
ls
- 执行以下命令,BWA中“mapping”对“fastq”文件进行比对。
- 生成字典。
gatk CreateSequenceDictionary -R human_g1k_v37.fasta -O human_g1k_v37.dict
回显显示如下红框中所示信息,表示执行完成。

生成的“dict”文件是为后面进行基因组的比对。
- 进行比对。
bwa mem -M -t 96 human_g1k_v37.fasta SRR742200_1.fastq SRR742200_2.fastq > SRR7.sam
表1 参数说明 参数
说明
-t
参数为线程数,一般与服务器核数一致。
回显显示如下红框中所示信息,表示执行完成。
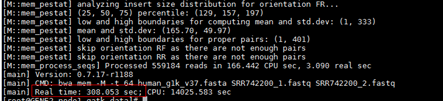
- 生成字典。
- 执行以下命令,Reorder对基因序列进行重新排序。
BWA生成的“sam”文件默认按字典式排序,需要转换为按染色体组型进行排序,由picard工具完成,GATK4中已集成picard功能。
gatk ReorderSam -I SRR7.sam -O SRR7_reorder.bam -R human_g1k_v37.fasta
回显显示如下红框中所示信息,表示执行完成。
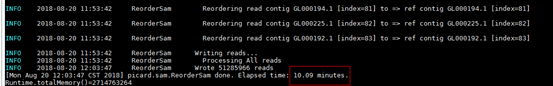
- Sort对基因序列按照坐标顺序从大到小排序。
- Spark运行于HDFS文件系统中,在linux系统下不可见,执行以下命令将“SRR7_reorder.bam”文件传入HDFS文件系统。
hadoop fs -mkdir /seqdata hadoop fs -put SRR7_reorder.bam /seqdata
- 执行以下命令确认“sparklog”目录已手动创建。
hadoop fs -mkdir /sparklog hadoop fs -ls /
- 执行以下命令进行排序。
gatk SortReadFileSpark -I hdfs://$(hostname):9000/seqdata/SRR7_reorder.bam -O hdfs://$(hostname):9000/seqdata/SRR7_sorted.bam -- --spark-runner SPARK --spark-master spark://$(hostname):7077

回显显示如下红框中所示信息,表示执行完成。

- 执行以下命令将生成的文件拷贝到数据目录。
hadoop fs -get /seqdata/SRR7_sorted.bam ./
- 执行以下命令查看数据目录。
ll
回显显示如下红框中所示信息,表示执行完成。

- Spark运行于HDFS文件系统中,在linux系统下不可见,执行以下命令将“SRR7_reorder.bam”文件传入HDFS文件系统。
- 执行以下命令,Add head对基因序列进行加头处理。
GATK2.0以上已不支持无头文件检测,该步骤可以在BWA比对时加-r进行,也可以使用GATK4中AddOrReplaceReadGroups工具完成。
gatk AddOrReplaceReadGroups -I SRR7_sorted.bam -O SRR7_header.bam -LB lib1 -PL illumina -PU unit1 -SM 20

回显显示如下红框中所示信息,表示执行完成。

- Mark Duplicates对基因序列去重。
- 执行以下命令将加头文件传入HDFS文件系统。
hadoop fs -put SRR7_header.bam /seqdata
- 执行以下命令查看HDFS文件系统。
hadoop fs -ls /seqdata

- 执行以下命令进行去重操作。
gatk MarkDuplicatesSpark -I hdfs://$(hostname):9000/seqdata/SRR7_header.bam -O hdfs://$(hostname):9000/seqdata/SRR7_markdup.bam -- --spark-runner SPARK --spark-master spark://$(hostname):7077

回显显示如下红框中所示信息,表示执行完成。

- 执行以下命令将加头文件传入HDFS文件系统。
- BQSR进行重新校准。
- 执行以下命令将原始文件传入HDFS文件系统。
hadoop fs -put dbsnp132_20101103.vcf /seqdata hadoop fs -put human_g1k_v37.fasta.2bit /seqdata hadoop fs -ls /seqdata
回显显示如红框中所示信息,表示执行完成。

- 执行BQSR(Base Quality Score Recalibration)。
gatk BQSRPipelineSpark -I hdfs://$(hostname):9000/seqdata/SRR7_markdup.bam -R hdfs://$(hostname):9000/seqdata/human_g1k_v37.fasta.2bit -O hdfs://$(hostname):9000/seqdata/SRR7_bqsr.bam --known-sites hdfs://$(hostname):9000/seqdata/dbsnp132_20101103.vcf --disable-sequence-dictionary-validation true -- --spark-runner SPARK --spark-master spark://$(hostname):7077

显示如下红框中所示信息表示执行完成。

- 执行以下命令将原始文件传入HDFS文件系统。
- 执行以下命令,进行HaplortypeCaller变异流程检测,生成变异体“vcf”文件。
gatk HaplotypeCallerSpark -R hdfs://$(hostname):9000/seqdata/human_g1k_v37.fasta.2bit -I hdfs://$(hostname):9000/seqdata/SRR7_bqsr.bam -O hdfs://$(hostname):9000/seqdata/SRR7.snp.raw.vcf -- --spark-runner SPARK --spark-master spark://$(hostname):7077


最终生成的数据信息如下所示。

