ARM平台使用Bioconductor软件包方法
介绍
- Bioconductor是一个基于R语言的开源生物信息学软件平台,旨在为生物学家和生物信息学家提供高质量、高效的工具和资源,以支持生物信息学研究。
- Bioconductor社区由一群来自世界各地的生物信息学家、统计学家和计算机科学家组成,他们致力于开发和维护生物信息学软件包、数据集和文档,以满足生物信息学研究的需求。 Bioconductor社区的软件包涵盖了各种生物信息学应用领域,包括基因表达分析、蛋白质组学、代谢组学、基因组学、转录组学、单细胞分析等。这些软件包提供了各种算法和工具,包括数据预处理、统计分析、可视化、机器学习等,可以帮助研究人员从海量的生物数据中提取有用的信息。
- Bioconductor社区还提供了丰富的数据资源,包括基因表达数据、蛋白质组数据、代谢组数据、基因组数据等,这些数据可以用于生物信息学研究和算法开发。
- Bioconductor社区的成员积极参与各种生物信息学研究项目,包括癌症研究、药物研发、基因组学研究等,他们不断开发和改进生物信息学工具和资源,以推动生物信息学研究的发展。
- Bioconductor官网:https://www.bioconductor.org/。
- Bioconductor目前已集合约2230生物信息领域软件包,最新的版本是3.17,对应的R版本是4.3。
- Bioconductor查询最新release包的地址:http://www.bioconductor.org/packages/release/BiocViews.html#___Software。
安装依赖包
安装Bioconductor软件包前提需要安装R语言,部分Bioconductor包还需要基于系统依赖库,例如bzip2等,一般推荐使用yum直接安装。
如下操作以R 4.3.0、Kylin V10为例,其余版本安装步骤类似。
- 使用PuTTY工具,以root用户登录服务器。
- 使用yum安装相关依赖。
yum install libXt-devel libX11-devel readline-devel pcre2-devel libcurl-devel libtiff-devel libpng-devel libjpeg-turo-devel cairo-devel freetype-devel harfbuzz-devel pango-devel zlib-devel
- 下载安装包并解压。
wget https://cran.r-project.org/src/base/R-4/R-4.3.0.tar.gz tar xvf R-4.3.0.tar.gz
- 编译安装R语言。
cd R-4.3.0 ./configure --prefix=/path/to/R_install make -j && make install

“/path/to”表示R语言的安装路径,用户安装时请以实际路径为准。
- 设置环境变量。
export PATH=/path/to/R_install/bin:$PATH export LD_LIBRARY_PATH=/path/to/R_install/lib/R/library:$LD_LIBRARY_PATH
安装Bioconductor
安装完R语言后,需要安装BiocManager管理器作为安装Bioconductor包的入口。
- 安装BiocManager。
- 打开R语言解析器。
R
- 安装BiocManager管理器。
install.packages("BiocManager")首次安装需要选择CRAN的镜像,推荐最近城市的镜像,以24:shenzhen举例。当服务器中出现“DONE(BiocManager)”,表示BiocManager管理器已安装完成。
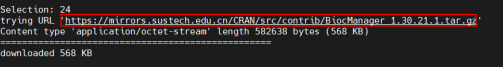
BiocManager的版本会根据当前R语言的版本进行匹配自动安装。
- 打开R语言解析器。
- 安装Bioconductor软件包。
Bioconductor支持自动与离线安装两种方式,推荐使用自动安装方式安装。如下操作以ACME为例,其余软件包安装步骤类似。
- 自动安装方式(适用于服务器已接通网络)
BiocManager::install("ACME")当服务器中出现“DONE(ACME)”,表示ACME已经安装完成。
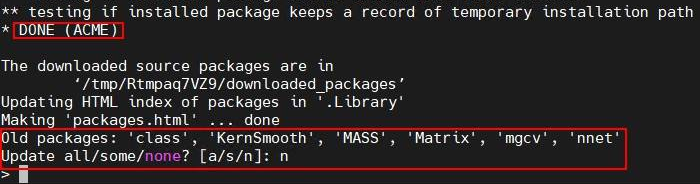
当最后提示“Old packages: XXX”时,请输入n不进行更新包操作,避免产生兼容性冲突。
- 离线安装方式(适用于依赖少的包和服务器无法接通网络)
- 打开ACME最新Release包地址http://www.bioconductor.org/packages/release/bioc/html/ACME.html,下载ACME的R源码包“ACME_2.56.0.tar.gz”并上传至服务器。
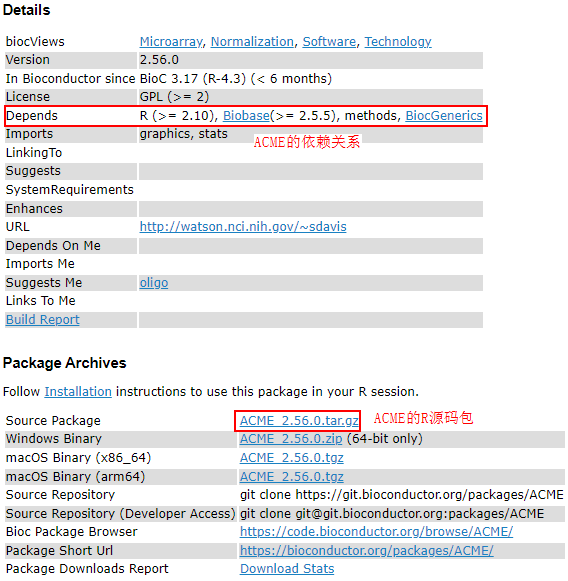
- 安装ACME。
R CMD INSTALL ACME_2.56.0.tar.gz
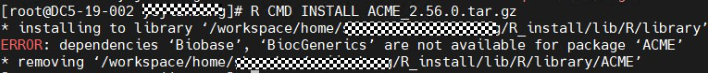
使用离线安装方式,在首次安装过程中一般会提示缺少某依赖,按照上述方法进行自动安装或离线安装缺少的依赖即可。例如以上报错为R环境中缺少Biobase和BiocGenerics包,下载Biobase和BiocGenerics包并上传至服务器,由于Biobase依赖BiocGenerics,则先安装BiocGenerics。
- 安装BiocGenerics。
R CMD INSTALL BiocGenerics_0.46.0.tar.gz
当服务器中出现“DONE(BiocGenerics)”,表示BiocGenerics已安装完成。
- 安装Biocbase。
R CMD INSTALL Biocbase_2.60.0.tar.gz
当服务器中出现“DONE(Biocbase)”,表示Biocbase已经安装完成。
- 安装BiocGenerics。
- 再次安装ACME。
R CMD INSTALL ACME_2.56.0.tar.gz
- 打开ACME最新Release包地址http://www.bioconductor.org/packages/release/bioc/html/ACME.html,下载ACME的R源码包“ACME_2.56.0.tar.gz”并上传至服务器。
- 自动安装方式(适用于服务器已接通网络)
Bioconductor软件使用方法
本章节以ACME举例,详细使用方法可参考ACME.R或者ACME.pdf,这2个文件来源于ACME的安装目录,一般是在“/path/to/R_install/lib/R/library/ACME/doc”,其余软件包使用方法同理,或到bioconductor包社区查询。
- 加载ACME库。
首先从两个nimblegen加载一些示例数据阵列,用于在ChiIP芯片实验中分析HOX基因。
library("ACME") datdir <- system.file('extdata',package='ACME') fnames <- dir(datdir) example.agff <- read.resultsGFF(fnames,path=datdir) example.agff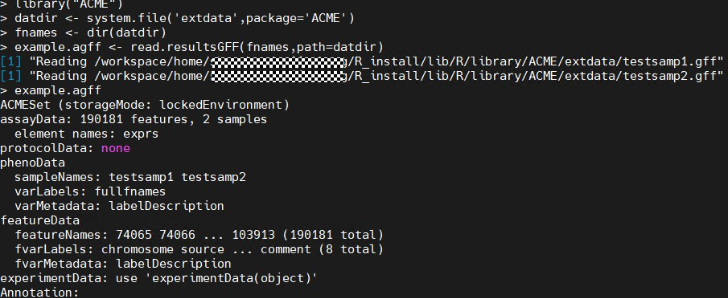
calc <- do.aGFF.calc(example.agff,window=1000,thresh=0.95)

- 作图和查找领域。
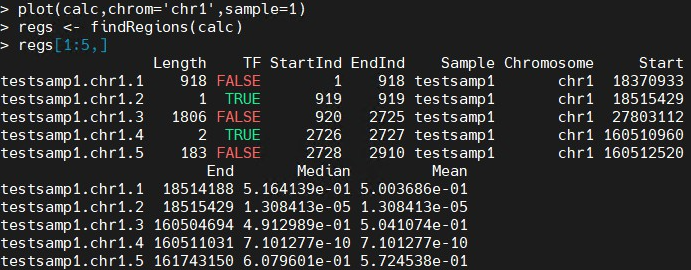
函数do.aGFF.calc将ACMESet对象作为输入,窗口大小(通常为2~3是实验中预期碎片大小的倍,并且足够大,至少包括大约10探针),以及将用于确定哪些探针被计数的阈值在卡方检验中为阳性。如果需要,结果可以在R图形窗口中绘制。原始信号通常是每个寡核苷酸(芯片/总基因组DNA)的数量将显示为灰点;相应的P值将以红色显示。虚线水平线表示在调用aGFF.calc中定义的阈值。在以下示例中,R绘制了来自1号染色体上任意选择的区域,基因组坐标为10,000~50,000。
plot(calc,chrom='chr1',sample=1) regs <- findRegions(calc) regs[1:5,]

- 生成用于在基因组浏览器中查看的文件。
Affymetrix集成Genorne浏览器(IGB)是一个非常快速的跨平台(基于Java)基因组浏览器,可以以多种格式显示数据。通过生成“sgr”文件,以完全交互式的方式查看原始数据和计算的p值。一个简单的函数write.sgr将生成这样的文件,然后可以加载到浏览器。该函数还作为如何生成其他文件格式的模型。使用小的修改,可以生成其他格式。
- 同时写入计算值和原始数据。
write.sgr(calc)
- 只写计算的数据。
write.sgr(calc,raw=FALSE)

- 导出到UCSC基因组浏览器bedgraph格式。
write.bedGraph(calc)
- 同时写入计算值和原始数据。
- 退出R语言解释器,并在当前目录生成文件。
q()
“Save workspace image?”表示是否保存历史命令,y表示是,n表示否,c表示取消。
ls -l
R语言使用包常用命令
以“ACME”举例,其余软件包使用方法同理。
- 返回指定包的版本。
packageVersion("ACME") - 加载指定包。
library("ACME") - 查看已加载的包。
(.packages())
- 查询所有包所在库的路径 。
.libPaths()
- 卸除已加载的包。
detach("package:ACME") detach("package:ACME",unload=TRUE) detach是卸除已经加载的包,同等于在运行环境中暂时卸除包,不希望这个包被加载使用。一般来说,在包使用函数冲突,检验函数依赖时比较有用。detach并不是卸载/彻底删除该包。