运行和验证
BEAST2安装包中“nexus”目录下提供了数据集Primates.nex,通过“beauti”配置参数,最后生成“xml”文件。导入生成的“xml”文件,运行beast命令,生成3个文件:日志文件(*.log)、树文件(*.trees)、xml状态文件(*.state)。
操作步骤
- 使用PuTTY工具,以root用户登录服务器。
- 执行以下命令,进入“nexus”目录。
cd /path/to/BEAST/beast/examples/nexus
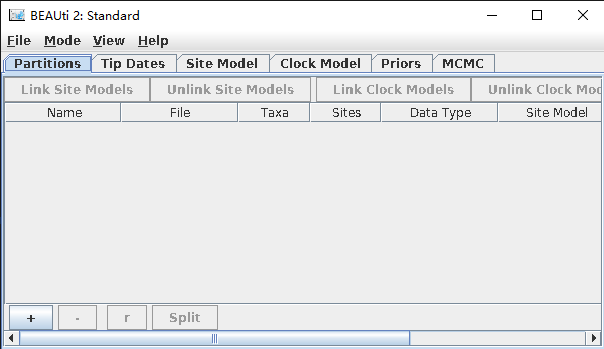
- 执行以下命令,弹出“BEAUti 2: Standard”弹窗,如图1所示。
beauti
- 序列导入 。
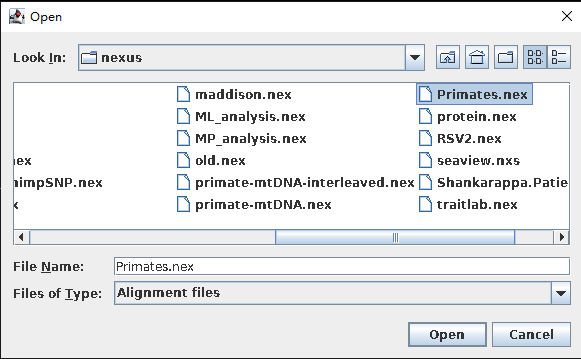
- Link/Unlink partition models(单击“Open”,弹出下列对话框)。
一般在分析中,导入的是比对好的基因序列信息,但有些情况下,也会存在partition情况。如果联合分析,就需要Link,否则不需要Unlink。

- 替代模型设定。
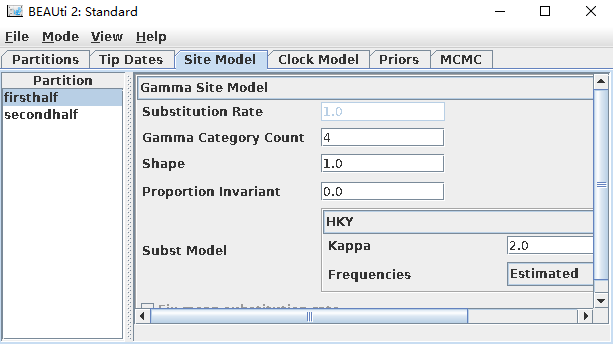
针对核甘酸替代模型,可供选择的有GTR、HKY、JC69、TN93,这里用HKY替代模型,Gamma Category Count(碱基替代速率变化等级)一般可设置为4-8,在这里选择4。其它参数如Substitution Rate(替代率)、Shape(GAMMA分布的形状参数)、Frequencies(碱基频率)在分析中均估计。
- 分子钟设置。
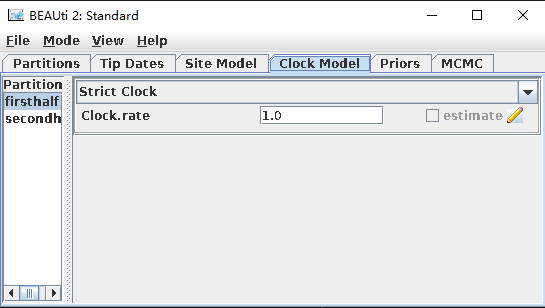
单击“Clock Model”,有Strict Clock(严格分子钟)、宽松分子钟(Relaxed Clock Exponential和Relaxed Clock Log Normal)等。 这里选择Strict Clock,不考虑模型中分子之间的速率变化差异。
- 先验证信息设定。
- 单击“Priors tree prior : Yule Model”,其它默认即可。
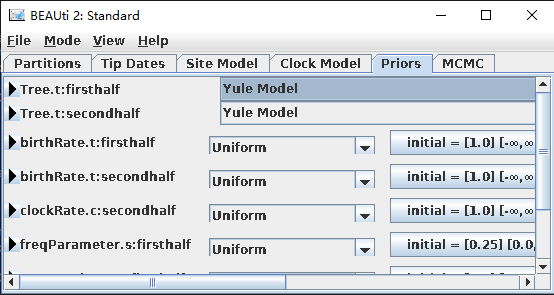
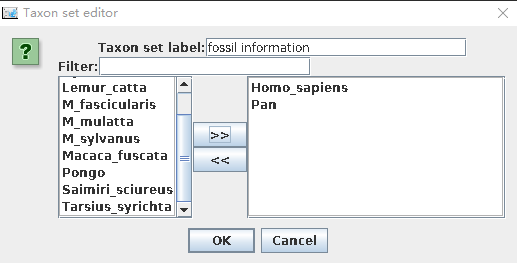
- 在Priors这一栏最下方有一个“+Add Prior”按钮, 单击确定后,弹出对话框,选择Tree.t:secondhalf。在Taxon set editor中自定义一个名字:如 fossil information,然后选择需要标定分歧时间的两个物种(如:Homo_sapiens、Pan),移到右边边框。
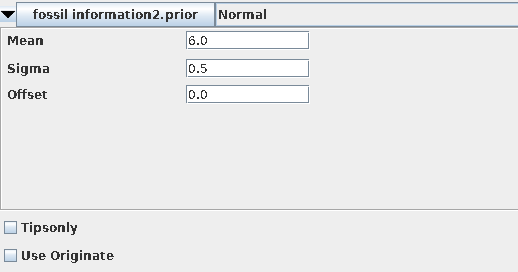
在校正节点上设置先验证分布信息,首先单击“monophyletic”下拉菜单([none])中选择Normal。随后设置分歧时间,Homo sapiens和Pan的分歧在5-7My,因此设定正态分布中心点是6My,标准偏差0.5My,并勾选“monophyletic”。
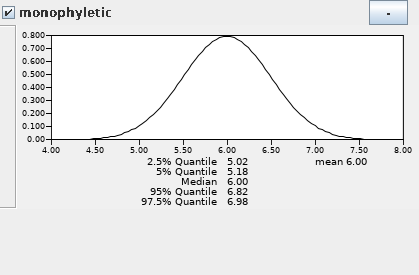
- 单击“Priors tree prior : Yule Model”,其它默认即可。
- MCMC选项设定(单击“MCMC”)。
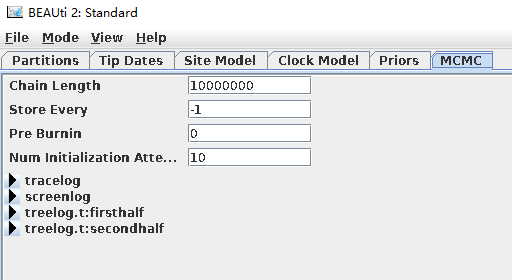
运行链长:10000000(默认1千万步)
tracelog:链长运行1000次抽样一次
screenlog: 运行1000次打印到屏幕一次
treelog.t:tree: 运行1000次打印一个拓扑结构树
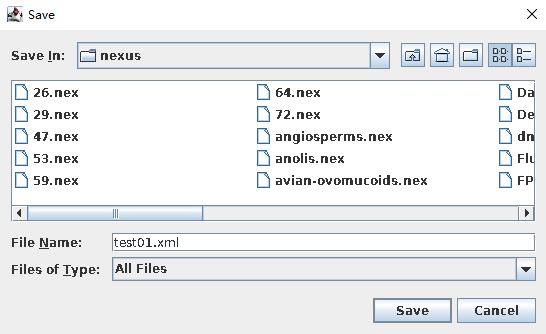
- “xml”文件保存。
单击“File—Save As”,定义一个输出文件名字,结果生成一个“xml”文件。
- 执行以下命令,进行beast测试。
beast -threads 128 test01.xml
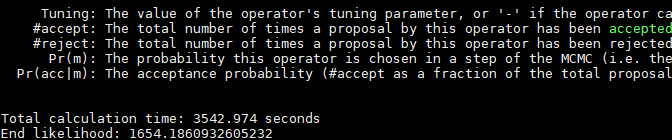
需要查看“Primates.log”日志中的“Total calculation time”数值,单位是“s”,数值越小性能越优。
输出的结果样例如下图所示。